18. Biology – phylogeny
18.1. Motivation, prerequisites, plan
18.1.1. Motivation
One of the most important areas of research in biology is that of phylogenetic analysis. This collection of techniques allows us to build an evolutionary tree showing how various species are related.
This type of analysis can also be used in other areas, such as tracing the origin of human spoken languages.
I find phylogenetic analysis to be fascinating because it gives us a sort of “webcam of the gods”, a view of the past (which we cannot see) which brought about the present state of things.
18.1.2. Prerequisites
The 10-hour “serious programming” course.
The “Data files and first plots” mini-course in Starting out - data files and first plots
Having the required libraries installed. Install them with:
$ sudo apt install python3-biopython python3-matplotlib
18.1.3. Plan
We will start by looking at a video tutorial of how to build a simple phylogenetic tree by hand. Then we will learn how to the biopython package to construct and visualize trees on that simple problem.
Then we discuss further projects in which we look for data sets to work with, including sets from our own genetic algorithm runs (where we also know the real evolutionary history), human languages (where we do not know the real history), and computer programming languages (where we should know most of the real history).
https://cnx.org/contents/24nI-KJ8@24.18:EmlvXoDL@7/Taxonomy-and-phylogeny
18.2. Start with a video and then make a simple table
Start with this Khan Academy tutorial on phylogenetic trees
Then we take their table of traits. Start with the empty table:
Species |
Feathers |
Fur |
Lungs |
Gizzard |
Jaws |
|---|---|---|---|---|---|
Lamprey |
|||||
Antelope |
|||||
Sea Bass |
|||||
Bald Eagle |
|||||
Alligator |
and write it on the board. Then fill out the tree on the board with the class. You can discuss what all these animals are, and look them up if necessary.
The table will end up looking like this:
Species |
Feathers |
Fur |
Lungs |
Gizzard |
Jaws |
|---|---|---|---|---|---|
Lamprey |
no |
no |
no |
no |
no |
Antelope |
no |
yes |
yes |
no |
yes |
Sea Bass |
no |
no |
no |
no |
yes |
Bald Eagle |
yes |
no |
yes |
yes |
yes |
Alligator |
no |
no |
yes |
yes |
yes |
Then use the principle of parsimony to create the phylogenetic tree, following the guidelines in the tutorial. The result should look like what you see in Figure 18.2.1.
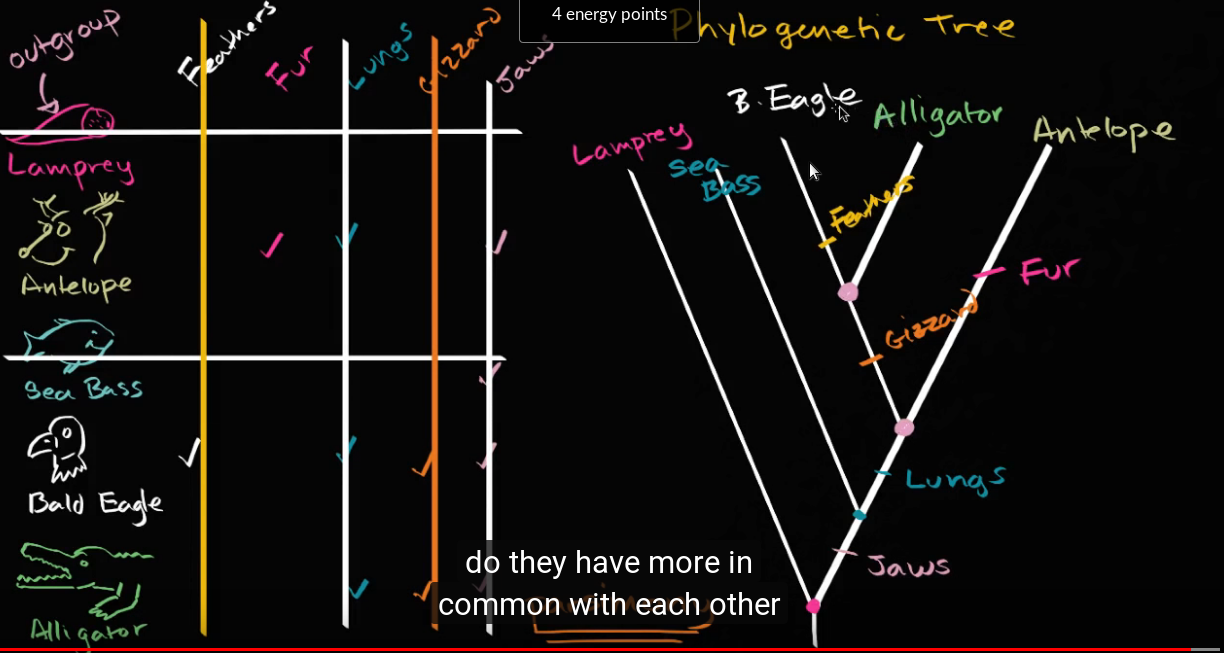
Figure 18.2.1 The resulting tree from the Khan Academy video example.
Discuss the meaning of parsimony as seen in this example. Connect it to Ockham’s razor.
18.3. Terminology
Clades, taxa, species, genotype, phenotype, …
The tree of life

Figure 18.3.1 Hillis’s tree of life based on completely sequenced genomes (from the Wikipedia image)
{kind=link}
18.4. First steps with biopython
Tutorials are at:
https://taylor-lindsay.github.io/phylogenetics/
and
http://biopython.org/DIST/docs/tutorial/Tutorial.html
The basic format of the library (or the part we will be using; biopython is HUGE) is below. You make a tree from some sort of text-based files (in this case just a string with some letters and parenthesis), then draw the tree. There are a lot of variations on this, but this is the fundemental structure.
from io import StringIO
from Bio import Phylo
from Bio.Phylo.TreeConstruction import DistanceCalculator
t = Phylo.read(StringIO("((a,b),c);" ), format="newick")
Phylo.draw(t)
18.5. Downloading the datasets
Data from opentreeoflife at:
https://tree.opentreeoflife.org/
I tied Streptococcus_mitis_NCTC_12261_ott725 at:
https://tree.opentreeoflife.org/opentree/argus/ottol@175918/Streptococcus
and downloaded the Newick format of the streptococcus subtree here.
with:
$ wget --output-document subtree-ottol-175918-Streptococcus.tre https://tree.opentreeoflife.org/opentree/default/download_subtree/ottol-id/175918/Streptococcus
And downloaded the opentreeoflife data with:
$ wget https://api.opentreeoflife.org/v3/study/ot_2221.tre
You can quickly visualize these datasets with:
import os
import matplotlib
import matplotlib.pyplot as plt
from Bio import Phylo
from Bio.Phylo.TreeConstruction import DistanceCalculator
tree = Phylo.read("ot_2221.tre", "newick")
Phylo.draw(tree)
tree = Phylo.read("subtree-ottol-175918-Streptococcus.tre", "newick")
Phylo.draw(tree)
Both are somewhat interesting to look ar
18.6. Preparing a tree by hand
Now let us prepare a tree where we input it ourselves. The format is
like what we saw in the example above: the tree is made with the call
Tree( "((a,b),c);" )
But we will make a slightly more interesting tree, the one we worked out in section Start with a video and then make a simple table. To do so enter the program in Listing 18.6.1.
#! /usr/bin/env python3
import matplotlib.pyplot as plt
from io import StringIO
from Bio import Phylo
from Bio.Phylo.TreeConstruction import DistanceCalculator
t = Phylo.read(StringIO("((((Eagle,Alligator),Antelope),Sea Bass),Lamprey);" ), format="newick")
Phylo.draw(t)
Phylo.draw(t, do_show=False)
plt.savefig('simple-tree.png')
print('Saved tree to simple-tree.png')
You can adjust the look of this tree. See the discussion in the second tutorial link in the first steps section. We can go through that and adjust our styles a bit and see how our tree looks. That being said, here is what the above program should output:
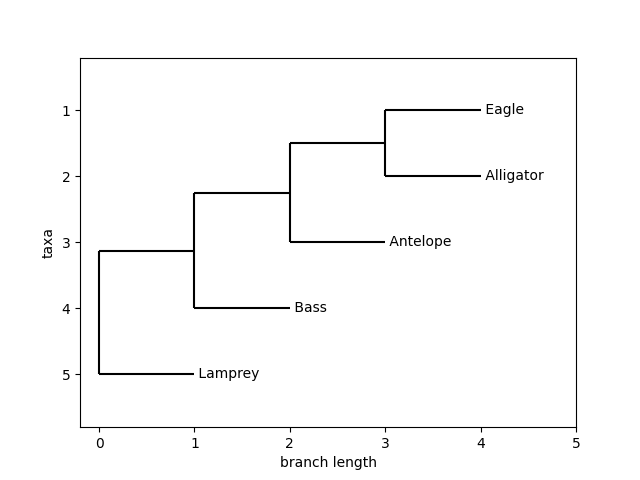
Figure 18.6.1 A version of the simple tree we deduced earlier.
18.7. Inferring a tree
The problem with the program in Listing 18.6.1 is that it prepares the tree, which you can view with your favorite PNG or SVG file viewer. But it does not find the tree. That is our next goal.
So we want to find the most likely evolutionary tree that would yield the result we see in the Table 18.2.2. This process is called inferring the phylogenetic tree from the table of characteristics.
To do this, we will use the .fa (or fasta) file format to encode the
information about traits. The format is fairly simple, with two lines
per animal: one with an arrow and the name, like >lamprey, and
then one with a single uppercase letter for each trait, like
NNNNN. When we put all five animals into a file together, it will
look something like:
>Lamprey
NNNNN
>Antelope
NYYNY
>Sea_Bass
NNNNY
>Bald_Eagle
YNYYY
>Alligator
NNYYY
along with a program to infer a tree from it:
#! /usr/bin/env python3
from Bio import Phylo
from Bio import AlignIO
from Bio.Phylo.TreeConstruction import DistanceTreeConstructor
from Bio.Phylo.TreeConstruction import DistanceCalculator
tree_alignment = AlignIO.read('simple-animals.fa', 'fasta')
calculator = DistanceCalculator('identity')
distance_matrix = calculator.get_distance(tree_alignment)
constructor = DistanceTreeConstructor(calculator, 'upgma')
tree = constructor.upgma(distance_matrix)
Phylo.draw(tree)
The output shows the same family links that we obtained by hand, but
the tree looks different because the root is placed differently. To
fix this, add tree.root_with_outgroup('Lamprey') directly before
the final line. The reason this works is it gives the computer a sense
of where to start, and creates the connections from there. If we
hadn’t included it, as you can see in the figure below, it would have
started from the point after the bass split off, giving a skewed view
of the tree.

Figure 18.7.1 A tree with all the same connections, but the wrong root.
After applying the fix, the tree should look like this:

Figure 18.7.2 The correctly inferred tree.
You can also view this tree as ascii by replacing the Phylo.draw(tree)
with Phylo.draw_ascii(tree).
We can save it to a file by adding a couple more lines:
import matplotlib.pyplot as plt
...
...
tree.root_with_outgroup('Lamprey')
Phylo.draw(tree, do_show=False)
plt.savefig('inferred_tree.png')
Phylo.draw_ascii(tree)
This will save a png formatted view of the tree, as well as showing you an ascii representation.
Now that we have a basic example out of the way, we are going to try a real-world example. This example is on variations a gene called CRAB across species, and can be copy-and-pasted from this link.
>crab_bovine ALPHA CRYSTALLIN B CHAIN (ALPHA(B)-CRYSTALLIN).
MDIAIHHPWIRRPFFPFHSPSRLFDQFFGEHLLESDLFPASTSLSPFYLR
PPSFLRAPSWIDTGLSEMRLEKDRFSVNLDVKHFSPEELKVKVLGDVIEV
HGKHEERQDEHGFISREFHRKYRIPADVDPLAITSSLSSDGVLTVNGPRK
QASGPERTIPITREEKPAVTAAPK
>crab_chicken ALPHA CRYSTALLIN B CHAIN (ALPHA(B)-CRYSTALLIN).
MDITIHNPLVRRPLFSWLTPSRIFDQIFGEHLQESELLPTSPSLSPFLMR
SPFFRMPSWLETGLSEMRLEKDKFSVNLDVKHFSPEELKVKVLGDMIEIH
GKHEERQDEHGFIAREFSRKYRIPADVDPLTITSSLSLDGVLTVSAPRKQ
SDVPERSIPITREEKPAIAGSQRK
>crab_human ALPHA CRYSTALLIN B CHAIN (ALPHA(B)-CRYSTALLIN).
MDIAIHHPWIRRPFFPFHSPSRLFDQFFGEHLLESDLFPTSTSLSPFYLR
PPSFLRAPSWFDTGLSEMRLEKDRFSVNLDVKHFSPEELKVKVLGDVIEV
HGKHEERQDEHGFISREFHRKYRIPADVDPLTITSSLSSDGVLTVNGPRK
QVSGPERTIPITREEKPAVTAAPK
>crab_mouse ALPHA CRYSTALLIN B CHAIN (ALPHA(B)-CRYSTALLIN) (P23).
MDIAIHHPWIRRPFFPFHSPSRLFDQFFGEHLLESDLFSTATSLSPFYLR
PPSFLRAPSWIDTGLSEMRLEKDRFSVNLDVKHFSPEELKVKVLGDVIEV
HGKHEERQDEHGFISREFHRKYRIPADVDPLAITSSLSSDGVLTVNGPRK
QVSGPERTIPITREEKPAVAAAPK
>crab_rabbit ALPHA CRYSTALLIN B CHAIN (ALPHA(B)-CRYSTALLIN).
MDIAIHHPWIRRPFFPFHSPSRLFDQFFGEHLLESDLFPTSTSLSPFYLR
PPSFLRAPSWIDTGLSEMRLEKDRFSVNLDVKHFSPEELKVKVLGDVIEV
HGKHEERQDEHGFISREFHRKYRIPADVDPLTITSSLSSDGVLTVNGPRK
QAPGPERTIPITREEKPAVTAAPK
>crab_rat ALPHA CRYSTALLIN B CHAIN (ALPHA(B)-CRYSTALLIN).
MDIAIHHPWIRRPFFPFHSPSRLFDQFFGEHLLESDLFSTATSLSPFYLR
PPSFLRAPSWIDTGLSEMRMEKDRFSVNLDVKHFSPEELKVKVLGDVIEV
HGKHEERQDEHGFISREFHRKYRIPADVDPLTITSSLSSDGVLTVNGPRK
QASGPERTIPITREEKPAVTAAPK
Note that you may have to trim the ends of the sequences to match the length; while this may loose some information contained in the sequences, it is small enough where the overall pattern will still show in the plot. We can reuse our tree-inferring program from earlier (make sure to change the file to the CRAB one and remove the line that sets the root), and it should produce something like Figure 18.7.3:

Figure 18.7.3 A graph showing the differences in the CRAB gene.
This graph shows a trend that makes a lot of sense: the mammals are all closely related, and the chicken is not closely related to them. In addition, the rat and mouse are closely related, which makes sense. The human in most closely related to the rabbit, and then the cow.
Biopython allowed us to learn all that information from meaningless (to us) sequences of letters. This can be incredibly useful for building phylogenetic trees, because you can simply plug in the genomes you are comparing and it will tell you how they are related. It’s not perfect, as we saw, and you may have to define an outgroup to “orient” the program. But other than that, it worked very well, and could build both or specifically engineered tree and a real example of a genome.
This is only a small taste of what biopython can do, and exploring it further would be reqrding for those with an interest in biology. The documentation and examples can be found here.
18.8. Other sequence analysis resources
Berkeley evolution course. 7 organisms and 7 features: https://evolution.berkeley.edu/evolibrary/article/phylogenetics_07
Cute with ladybugs, but just 6 elements and 7 features: https://bioenv.gu.se/digitalAssets/1580/1580956_fyltreeeng.pdf
Another video giving step-by-step for building a tree by hand: https://www.youtube.com/watch?v=09eD4A_HxVQ
18.9. Linguistics datasets
First discuss the issues of generating a string representation of language features. One example of the issues involved is given in this article:
https://brill.com/view/journals/ldc/3/2/article-p245_4.xml?lang=en
where they discuss how to align the English “horn” with the latin “kornu”. This then allows you to define a “genetic distance” between the same word in two different languages. One such measure is the “Levenshtein normalized distance” (LDN), which takes values between 0 and 1.
This can then be used with the ASJP (Automated Similarity Judgement Program) https://en.wikipedia.org/wiki/Automated_Similarity_Judgment_Program database which is based on a word list. The database is at https://asjp.clld.org/
Download the database from
https://asjp.clld.org/download
and look at the listss18.txt file and see how languages we know (English, Italian, Spanish, Russian) are represented.
Look at the results in WorldLanguageTree001.pdf
One oft-used list is the the Swadesh list https://en.wikipedia.org/wiki/Swadesh_list which has 100 terms. Some of these are “I”, “you”, “we”, “this”, “that”, “person”, “fish”, “dog”, “foot”, “hand”, “sun”, “mountain”, various basic colors, and so forth. There is also an abbreviated 35-word list. The ASJP uses a 40-word list, similar to the Swadesh list.
Each part of a word gets an ASJP code and an IPA (International Phonetic Alphabet) designation of how it’s pronounced.
18.9.1. lingpy.org
Go through the tutorial, starting at:
http://www.lingpy.org/tutorial/index.html
install with
pip3 install lingpy
then simple examples at:
http://www.lingpy.org/examples.html
then the workflow tutorial at:
http://www.lingpy.org/tutorial/workflow.html
and the cookbook at:
18.9.2. elinguistics.com
Overall language evolutionary tree: http://www.elinguistics.net/Language_Evolutionary_Tree.html You can follow the links to some detailed discussion of timelines at http://www.elinguistics.net/Language_Timelines.html as well as in-depth discussion of encoding and language comparison. In particular you will find various sounds for key encoding words at http://www.elinguistics.net/Compare_Languages.aspx?Language1=English&Language2=German&Order=Details
The choice of “basis words” to use as “genetic markers” is described at http://www.elinguistics.net/Lexical_comparison.html and the continuing pages http://www.elinguistics.net/Sound_Correspondence.html and back to the example of English to German at http://www.elinguistics.net/Compare_Languages.aspx?Language1=English&Language2=German&Order=Details
18.9.3. Others
https://en.wikipedia.org/wiki/Tree_model#Perfect_phylogenies
https://en.wikipedia.org/wiki/Tree_model
https://en.wikipedia.org/wiki/Language_family
https://en.wikipedia.org/wiki/Tree_model#CITEREFNakhleh2005
https://www.theguardian.com/education/gallery/2015/jan/23/a-language-family-tree-in-pictures
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3049109/
https://glottolog.org/resource/languoid/id/stan1293
https://glottolog.org/glottolog/family
https://www.ethnologue.com/browse/families
https://glottolog.org/resource/languoid/id/macr1271
https://science.sciencemag.org/content/323/5913/479
Look at the PDF for this paper on phylogeny of polynesian languages:
And this one with very nice-looking pictures of Japonic language evolution and some discussion of word differentiation.
https://royalsocietypublishing.org/doi/full/10.1098/rspb.2011.0518
Natural language processing with Python and NLTK:
18.10. Evolution of programming languages
https://www.i-programmer.info/news/98-languages/8809-the-evolution-of-programming-languages.html
https://royalsocietypublishing.org/doi/full/10.1098/rsif.2015.0249